hicMergeDomains¶
hicMergeDomains takes as input multiple TAD domain files from hicFindTads. It merges TADs from different resolutions to one TAD domains file, considers protein peaks from known TAD binding sites and computes a dependency graph of the TADs.
Two TADs are considered as one if they don’t overlap at x bins given by –value; TAD borders need to match the protein peaks given by –proteinFile; a relation between two TADs is given by their overlap of area in percent, parameter –percent. The protein peaks are only considered if in one bin at least –minPeak.
An example usage is:
$ hicMergeDomains –domainFiles 10kbtad_domains.bed 50kbtad_domains.bed –proteinFile ctcf_sorted.bed –outputMergedList two_files_ctcf –outputRelationList two_files_relation_ctcf –outputTreePlotPrefix two_files_plot_ctcf –outputTreePlotFormat pdf
usage: hicMergeDomains --domainFiles DOMAINFILES [DOMAINFILES ...]
[--proteinFile PROTEINFILE]
[--minimumNumberOfPeaks MINIMUMNUMBEROFPEAKS]
[--value VALUE] [--percent PERCENT]
[--outputMergedList OUTPUTMERGEDLIST]
[--outputRelationList OUTPUTRELATIONLIST]
[--outputTreePlotPrefix OUTPUTTREEPLOTPREFIX]
[--outputTreePlotFormat OUTPUTTREEPLOTFORMAT] [--help]
[--version]
Required arguments¶
- --domainFiles, -d
The domain files of the different resolutions is required
Optional arguments¶
- --proteinFile, -p
In order to be able to better assess the relationship between TADs, the associated protein file (e.g. CTCF for mammals) can be included. The protein file is required in broadpeak format
- --minimumNumberOfPeaks, -m
Optional parameter to adjust the number of protein peaks when adapting the resolution to the domain files. At least minimumNumberOfPeaks of unique peaks must be in a bin to considered. Otherwise the bin is treated like it has no peaks (Default: 1).
Default: 1
- --value, -v
Determine a value by how much the boundaries of two TADs must at least differ to consider them as two separate TADs (Default: 5000).
Default: 5000
- --percent, -pe
For the relationship determination, a percentage is required from which area coverage the TADs are related to each other.For example, a relationship should be entered from 5 percent area coverage -p 0.05 (Default: 0.5).
Default: 0.5
- --outputMergedList, -om
File name for the merged domains list (Default: “mergedDomains.bed”).
Default: “mergedDomains.bed”
- --outputRelationList, -or
File name for the relationship list of the TADs (Default: “relationList.txt”).
Default: “relationList.txt”
- --outputTreePlotPrefix, -ot
File name prefix for the relationship tree of the TADs (Default: “relationship_tree_”).
Default: “relationship_tree_”
- --outputTreePlotFormat, -of
File format of the relationship tree. Supported formats are listed on: https://www.graphviz.org/doc/info/output.html (Default: “pdf”).
Default: “pdf”
- --version
show program’s version number and exit
This tool is the result of the bachelor thesis (in German) from Sarah Domogalla at the Bioinformatics lab of the Albert-Ludwigs-University Freiburg. The thesis was written in winter semester 2019/2020.
The thesis covers the issue of differently detected TADs of the same data but on different resolutions.
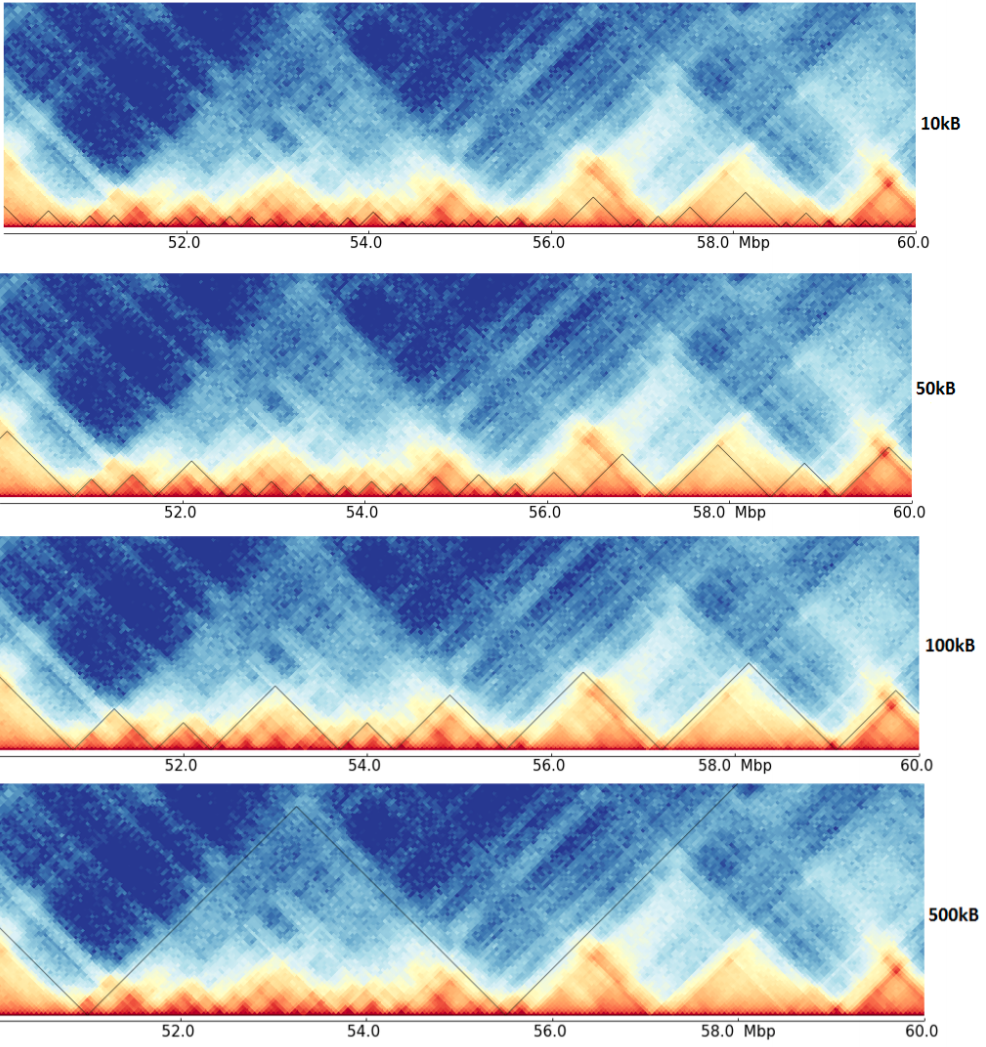
As it can be seen here, the detected TADs on the four resolutions are quite different, and have maybe a hierarchy. Compare for example the region around 53 Mb in the different resolutions, it is unclear if these are multiple small TADs as suggest by the 10kb resolution, mid-size one as seen by 50 kb or one big, as detected on 100 kb resolution. Consider the next image for an abstract representation of this problem.
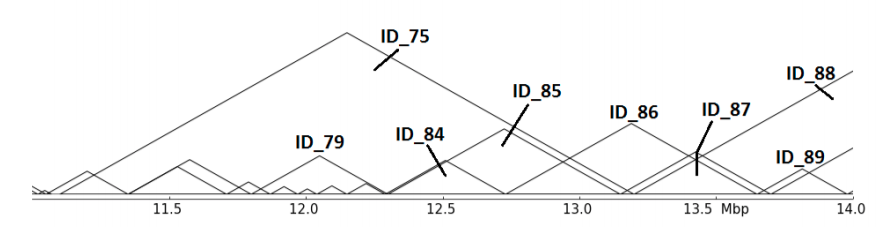
The abstract representation shows multiple issues: Are ID_85,ID_86 and ID_87 related in any hierarchy? Which role plays ID_75? hicMergeDomains tries to solve this by providing the parameters –value and –percentage to control how the relationship is determined and if TADs are duplicated and therefore deleted.
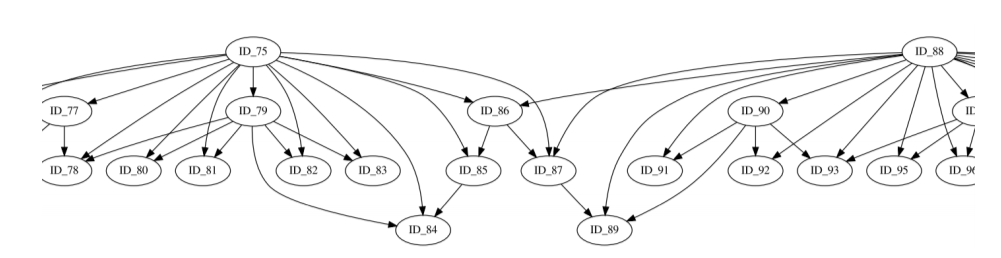
This abstract representation of the hierarchical TADs is created for each chromosome and provides insights for the organization of the chromatin structure in the three dimensional space.
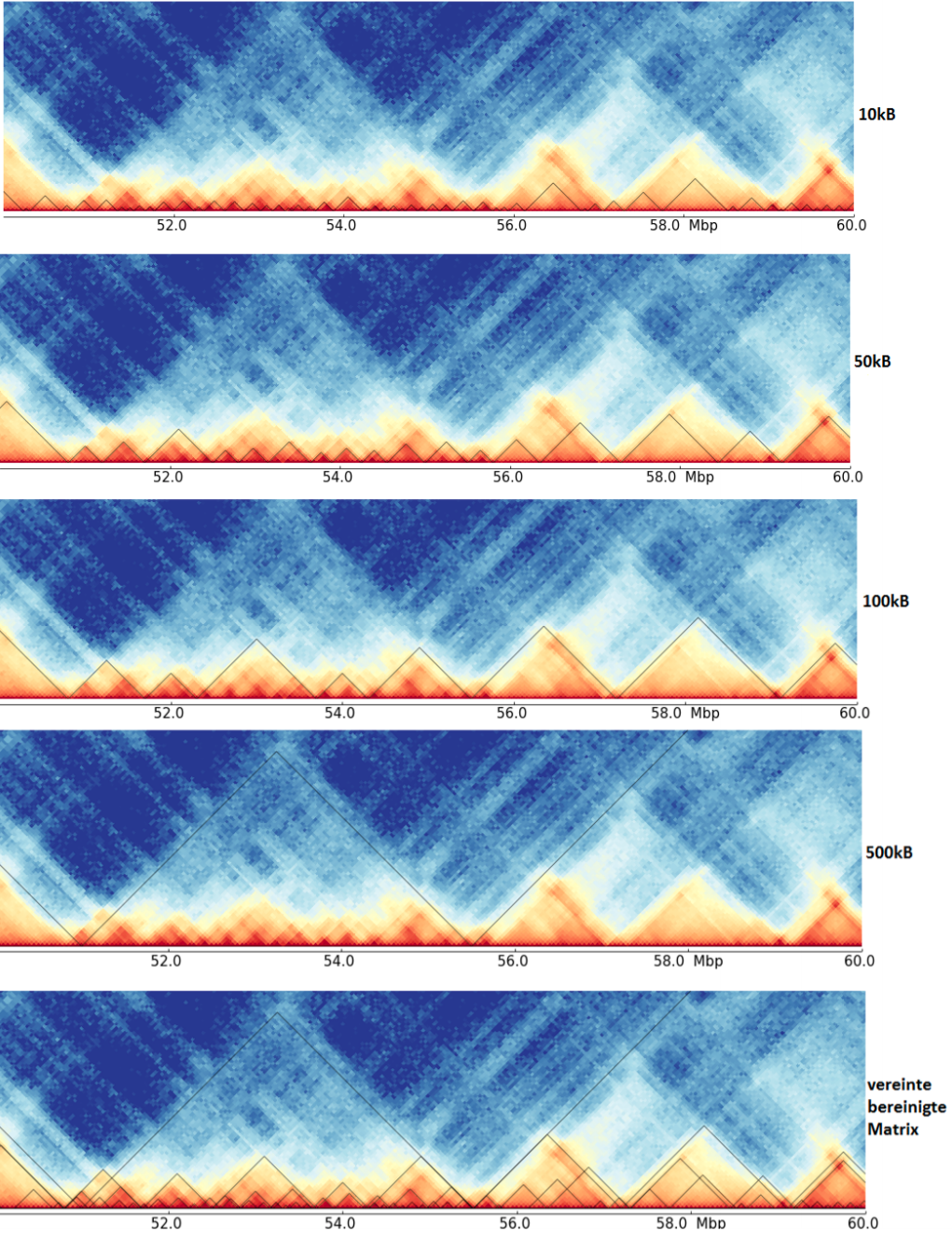
The last image shows a cleaned TAD detection based on all four inputs.