hicDifferentialTAD¶
Computes differential TADs by comparing the precomputed TAD regions of the target matrix with the same regions of the control matrix. Please notice that the matrices need to have the same read coverage, this can be achieved with hicNormalize and the ‘smallest’-mode. H0 is the assumption that two regions are identical, the rejected files contain therefore the as differential considered regions.
usage: hicDifferentialTAD [--targetMatrix TARGETMATRIX]
[--controlMatrix CONTROLMATRIX]
[--tadDomains TADDOMAINS]
[--outFileNamePrefix OUTFILENAMEPREFIX]
[--pValue PVALUE]
[--mode {intra-TAD,left-inter-TAD,right-inter-TAD,all}]
[--modeReject {all,one}] [--threads THREADS]
[--help] [--version]
Required arguments¶
- --targetMatrix, -tm
The matrix which was used to compute the TADs
- --controlMatrix, -cm
The control matrix to test the TADs for a differential interaction pattern.
- --tadDomains, -td
The TADs domain file computed by hicFindTADs.
- --outFileNamePrefix, -o
Outfile name prefix to store the accepted / rejected H0 TADs.
Default: “output_differential_tad”
Optional arguments¶
- --pValue, -p
H0 is considered as ‘two regions are identical.’ i.e. all regions with a test result of <= p-value are rejected and considered as differential (Default: 0.05).
Default: 0.05
- --mode, -m
Possible choices: intra-TAD, left-inter-TAD, right-inter-TAD, all
Consider only intra-TAD interactions, or additional left inter-TAD, right inter-TAD or all (Default: “all”).
Default: “all”
- --modeReject, -mr
Possible choices: all, one
All test of a mode must be rejected (all) or reject region (and accept it is differential) as soon as at least one region is having a p-value <= –pValue (Default: “one”).
Default: “one”
- --threads, -t
Number of threads to use, the parallelization is implemented per chromosome (Default: 4).
Default: 4
- --version
show program’s version number and exit
hicDifferentialTAD computes with a treatment Hi-C file, a control Hi-C file and a precomputed TAD domains file if the detected TADs are differential between the treatment and the control sample. The TADs need to be precomputed on the treatment file with _hicFindTADs.
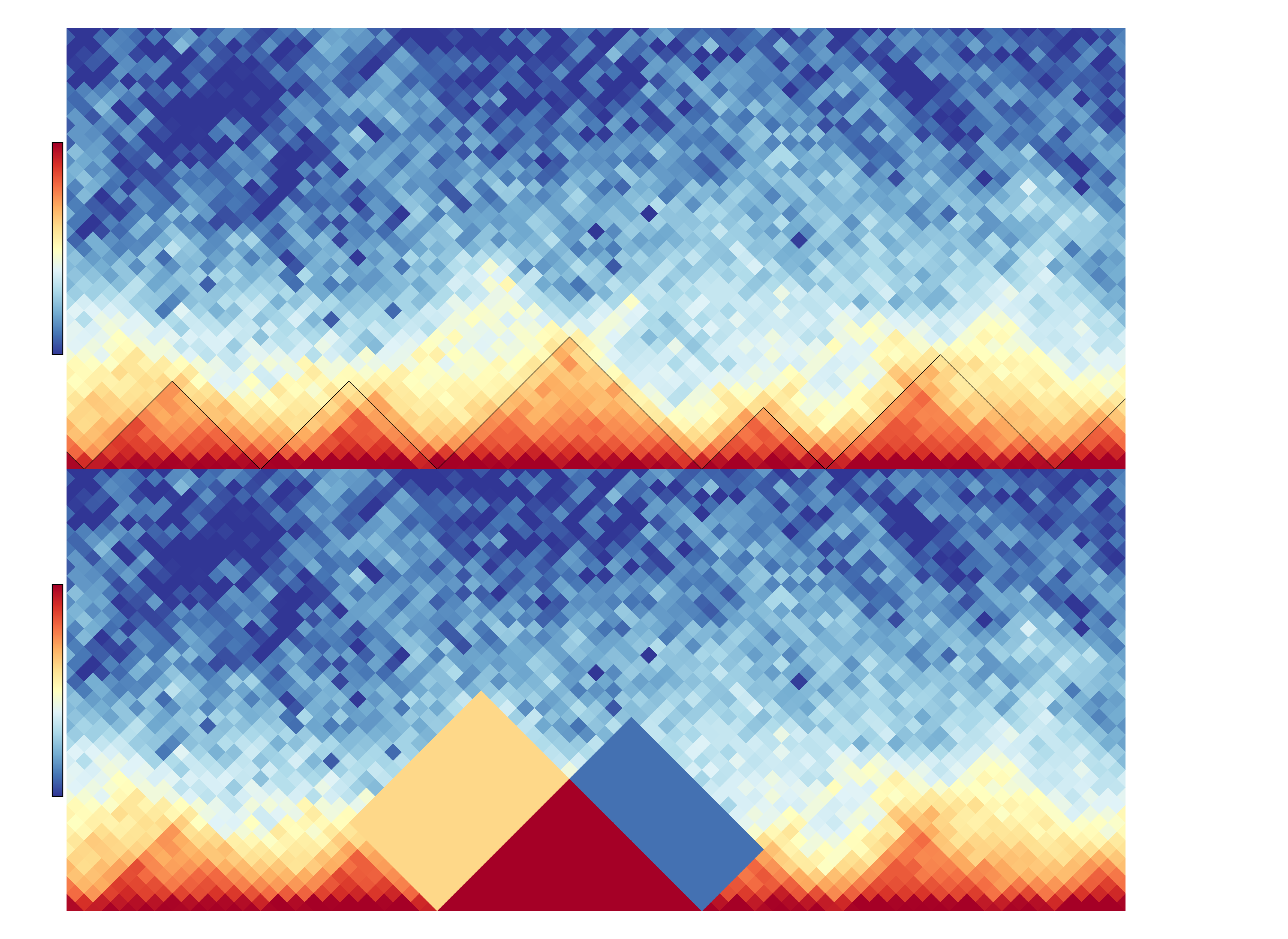
hicDifferentialTAD extract per TAD three regions: the intra-TAD, the left and right inter-TAD region. In the following image, the upper visualization shows a region with the detected TADs which are indicated by the black lines. The bottom shows as an example which regions are used for the differential test: the intra-TAD region is highlighted in red, the left inter-TAD in sandy-color and the right inter-TAD in blue. Between two samples a Wilcoxon rank-sum test is applied for a TAD under H0: ‘The regions are equal’. For all three regions of a TAD the rank-sum test is independently applied. The user has the choice with the two parameters ‘mode’ and ‘modeReject’ to define if a) all three regions should be considered (‘all’), or only a subset e.g. the intra-TAD or intra-TAD and left inter-TAD should be considered; and b) if all regions need to have lower p-value than the user given to reject H0 or if it is enough that at least one region is rejecting H0 to consider the region as differential.
Example usage¶
$ hicDifferentialTAD -tm GSM2644945_Untreated-R1.100000_chr1.cool -cm GSM2644947_Auxin2days-R1.100000_chr1.cool -td untreated_R1_domains.bed -o differential -p 0.01 -t 4 -mr all
In this example data from Nora et al. “Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization”, Cell 169.5 (2017): 930-944 is used [GEO: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE98671]
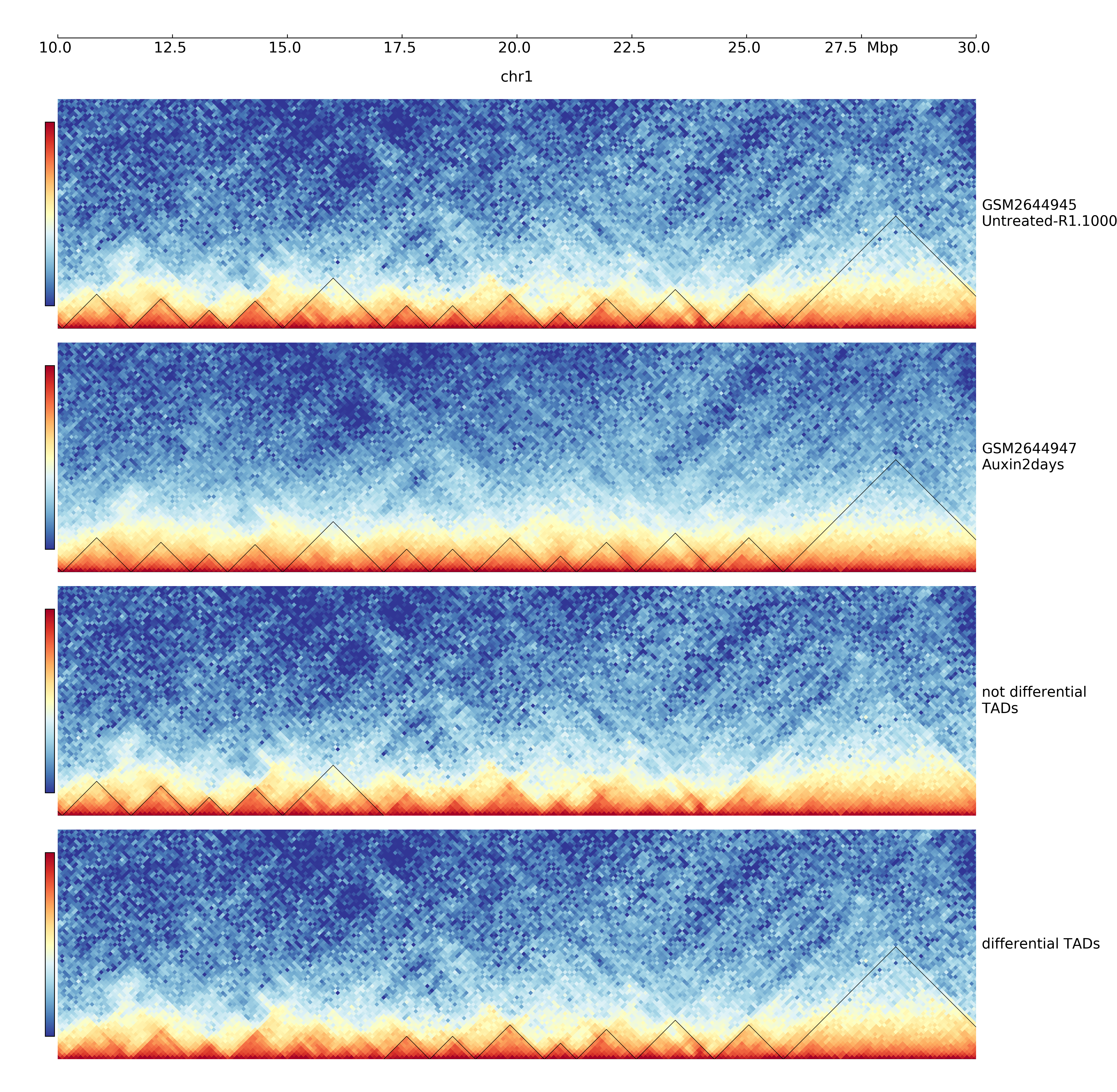
The output are two BED-similar files: the ‘_accepted.diff_tad’ and ‘_rejected.diff_tad’ file. The difference to a real BED file is a) the usage of a header starting with ‘#’, the first six columns are BED6 standard, however, there are three additional columns with the p-values of each intra-TAD, left-inter-TAD and right-inter-TAD test. The score value and name is copied from the domains.bed file which is a output of hicFindTADs.
# Created with HiCExplorer's hicDifferentialTAD version 3.5-dev
# H0 'regions are equal' H0 is rejected for all p-value smaller or equal the user given p-value threshold; i.e. regions in this file are considered as differential.
# Rejected regions with Wilcoxon rank-sum test to p-value: 0.01 with used mode: all and modeReject: all
# Chromosome start end name score strand p-value left-inter-TAD p-value right-inter-TAD p-value intra-TAD
chr1 4400000 6200000 ID_0.01_1 -0.5630275 . nan 0.42772134044376386 0.0001942482956610518
chr1 6200000 7300000 ID_0.01_2 -0.235798 . 0.3479058102348157 0.651801360704674 0.011174626122333891
chr1 7300000 9500000 ID_0.01_3 -0.44334 . 0.7619708385959597 0.9408966423531526 4.668547072331386e-06
chr1 10100000 11600000 ID_0.01_5 -0.6844015 . 0.10355136240139871 0.2022260523997077 0.006197912102328514
chr1 11600000 12900000 ID_0.01_6 -0.607587 . 0.39564035674322084 0.04934747494654432 5.069275897797787e-07
chr1 12900000 13700000 ID_0.01_7 -0.6303795 . 0.030328896271663634 0.048097680638217614 0.0023999817017426378
chr1 13700000 14900000 ID_0.01_8 -1.2063985 . 0.07605786424686575 0.5322636852613494 1.8146100883843892e-06
chr1 14900000 17100000 ID_0.01_9 -0.3406565 . 0.4982863585572014 0.692745144471043 0.002562121461829293
chr1 17100000 18100000 ID_0.01_10 -0.230354 . 0.8237750360973404 0.011911117420279341 0.00020602838341698515
chr1 18100000 19100000 ID_0.01_11 -0.5135365 . 0.007089417650089524 0.07553560212368073 0.03561337274424189