hicPlotMatrix¶
Creates a heatmap of a Hi-C matrix.
usage: hicPlotMatrix --matrix MATRIX --outFileName OUTFILENAME [--title TITLE]
[--scoreName SCORENAME] [--perChromosome]
[--clearMaskedBins]
[--chromosomeOrder CHROMOSOMEORDER [CHROMOSOMEORDER ...]]
[--region REGION] [--region2 REGION2] [--log1p] [--log]
[--colorMap COLORMAP] [--vMin VMIN] [--vMax VMAX]
[--dpi DPI] [--bigwig BIGWIG [BIGWIG ...]]
[--bigwigAdditionalVerticalAxis]
[--vMinBigwig VMINBIGWIG] [--vMaxBigwig VMAXBIGWIG]
[--flipBigwigSign]
[--scaleFactorBigwig SCALEFACTORBIGWIG]
[--fontsize FONTSIZE] [--rotationX ROTATIONX]
[--rotationY ROTATIONY]
[--increaseFigureWidth INCREASEFIGUREWIDTH]
[--increaseFigureHeight INCREASEFIGUREHEIGHT]
[--loops LOOPS]
[--loopLargeRegionsOperation {first,last,center}]
[--tads TADS] [--help] [--version]
Required arguments¶
- --matrix, -m
Path of the Hi-C matrix to plot.
- --outFileName, -out
File name to save the image.
Optional arguments¶
- --title, -t
Plot title.
- --scoreName, -s
Score name label for the heatmap legend.
- --perChromosome
Instead of plotting the whole matrix, each chromosome is plotted next to the other. This parameter is not compatible with –region.
Default: False
- --clearMaskedBins
If set, masked bins are removed from the matrix and the nearest bins are extended to cover the empty space instead of plotting black lines.
Default: False
- --chromosomeOrder
Chromosomes and order in which the chromosomes should be plotted. This option overrides –region and –region2.
- --region
Plot only this region. The format is chr:start-end The plotted region contains the main diagonal and is symmetric unless –region2 is given.
- --region2
If given, then only the region defined by –region and –region2 is given. The format is the same as –region1.
- --log1p
Plot the log1p of the matrix values.
Default: False
- --log
Plot the MINUS log of the matrix values.
Default: False
- --colorMap
Color map to use for the heatmap. Available values can be seen here: http://matplotlib.org/examples/color/colormaps_reference.html (Default: “RdYlBu_r”).
Default: “RdYlBu_r”
- --vMin
Minimum score value.
- --vMax
Maximum score value.
- --dpi
Resolution for the image in case theoutput is a raster graphics image (e.g png, jpg) (Default: 72).
Default: 72
- --bigwig
Bigwig file to plot below the matrix. This can for example be used to visualize A/B compartments or ChIP-seq data.
- --bigwigAdditionalVerticalAxis
Add an additional axis to determine the values of a bigwig file in 2D better.
Default: False
- --vMinBigwig
Minimum score value for bigwig.
- --vMaxBigwig
Maximum score value for bigwig
- --flipBigwigSign
The sign of the bigwig values are flipped. Useful if hicPCA gives inverted values.
Default: False
- --scaleFactorBigwig
Scale the values of a bigwig file by the given factor (Default: 1.0).
Default: 1.0
- --fontsize
Fontsize in the plot for x and y axis (Default: 10).
Default: 10
- --rotationX
Rotation in degrees for the labels of x axis (Default: 0).
Default: 0
- --rotationY
Rotation in degrees for the labels of y axis (Default: 0).
Default: 0
- --increaseFigureWidth
Plotting additional bigwig tracks can cause compression in x or y direction of the heatmap. Set this factor to a value unequal to 0 to correct this (Default: 0.5).
Default: 0.5
- --increaseFigureHeight
Plotting additional bigwig tracks can cause compression in x or y direction of the heatmap. Set this factor to a value unequal to 0 to correct this (Default: 0.5).
Default: 0.5
- --loops
Bedgraph file to plot detected long range contacts from hicDetectLoops.
- --loopLargeRegionsOperation
Possible choices: first, last, center
If a given coordinate in the bed file is larger than a bin of the input matrix, by default only the first bin is taken into account. However there are more possibilities to handel such a case. Users can ask for the last bin or for center of the region.
Default: “first”
- --tads
Bedgraph file to plot detected tads
- --version
show program’s version number and exit
Details¶
hicplotMatrix takes a Hi-C matrix and plots the interactions of all or some chromosomes.
Examples¶
Here’s an example of Hi-C data from wild-type D. melanogaster embryos.
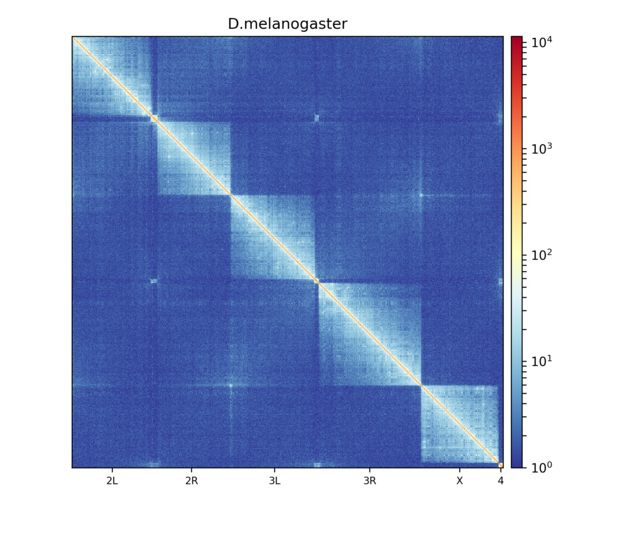
This plot shows all contacts of a Hi-C matrix. Its bins were merged into 25 kb bins using hicMergeMatrixBins. Alternatively, chromosomes can be plotted separately:
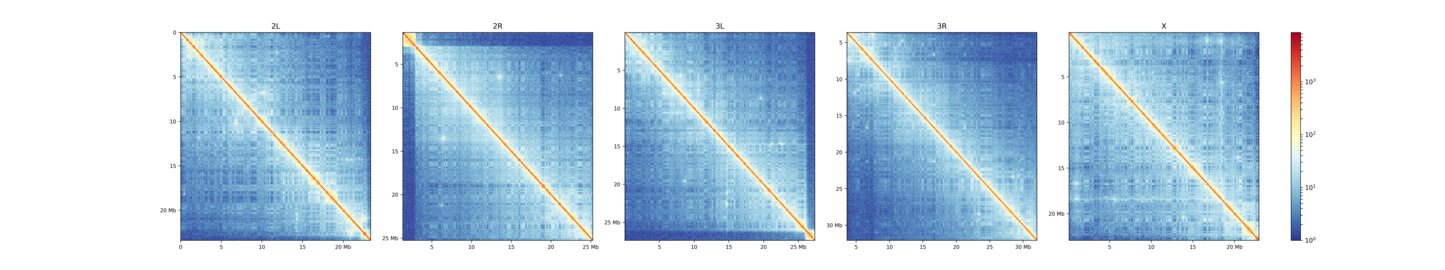
$ hicPlotMatrix -m Dmel.h5 -o hicPlotMatrix.png \
-t 'D.melanogaster (--perChromosome)' --log1p \
--clearMaskedBins --chromosomeOrder 2L 2R 3L 3R X --perChromosome