hicAverageRegions¶
Sums Hi-C contacts around given reference points and computes their average. This tool is useful to detect differences at certain reference points as for example TAD boundaries between samples.
WARNING: This tool can only be used with fixed bin size Hi-C matrices. No guarantees how and if it works on restriction site interaction matrices.
usage: hicAverageRegions --matrix MATRIX --regions REGIONS
(--range RANGE RANGE | --rangeInBins RANGEINBINS RANGEINBINS)
--outFileName OUTFILENAME [--help]
[--coordinatesToBinMapping {start,center,end}]
[--considerStrandDirection] [--version]
Named Arguments¶
- --range, -ra
Range of region up- and downstream of each region to include in genomic units.
- --rangeInBins, -rib
Range of region up- and downstream of each region to include in bin units.
Required arguments¶
- --matrix, -m
The matrix to use for the average of TAD regions.
- --regions, -r
BED file which stores a list of regions that are summed and averaged
- --outFileName, -o
File name to save the average regions TADs matrix.
Optional arguments¶
- --coordinatesToBinMapping, -cb
Possible choices: start, center, end
If the region contains start and end coordinates, define if the start, center (start + (end-start) / 2) or end bin should be used as start for range.This parameter is only important to set if the given start and end coordinates are not in the same bin (Default: “start”).
Default: “start”
- --considerStrandDirection
This parameter specifies if the strand information is taken into account for the aggregation. It has the effect that the contacts of a reverse strand region are inverted e.g. [1,2,3] becomes [3,2,1].
Default: False
- --version
show program’s version number and exit
hicAverageRegions takes as input a BED files with genomic positions, these are the reference points to sum up and average the regions up- and downstream of all these positions, good reference points are e.g. the borders of TADs. This can help to determine changes in the chromatin organization and TAD structure changes.
In the following example the 10kb resolution interaction matrix of Rao 2014 is used.
The first step computes the TADs for chromosome 1.
$ hicFindTADs -m GSE63525_GM12878_insitu_primary_10kb_KR.cool --outPrefix TADs
--correctForMultipleTesting fdr --minDepth 30000 --maxDepth 100000
--step 10000 -p 20 --chromosomes 1
Next, we use the domains.bed file of hicFindTADs to use the borders of TADs as reference points. As a range up- and downstream of each reference point 100kb are chosen.
$ hicAverageRegions -m GSE63525_GM12878_insitu_primary_10kb_KR.cool
-r TADs_domains.bed --range 100000 100000 --outFileName primary_chr1
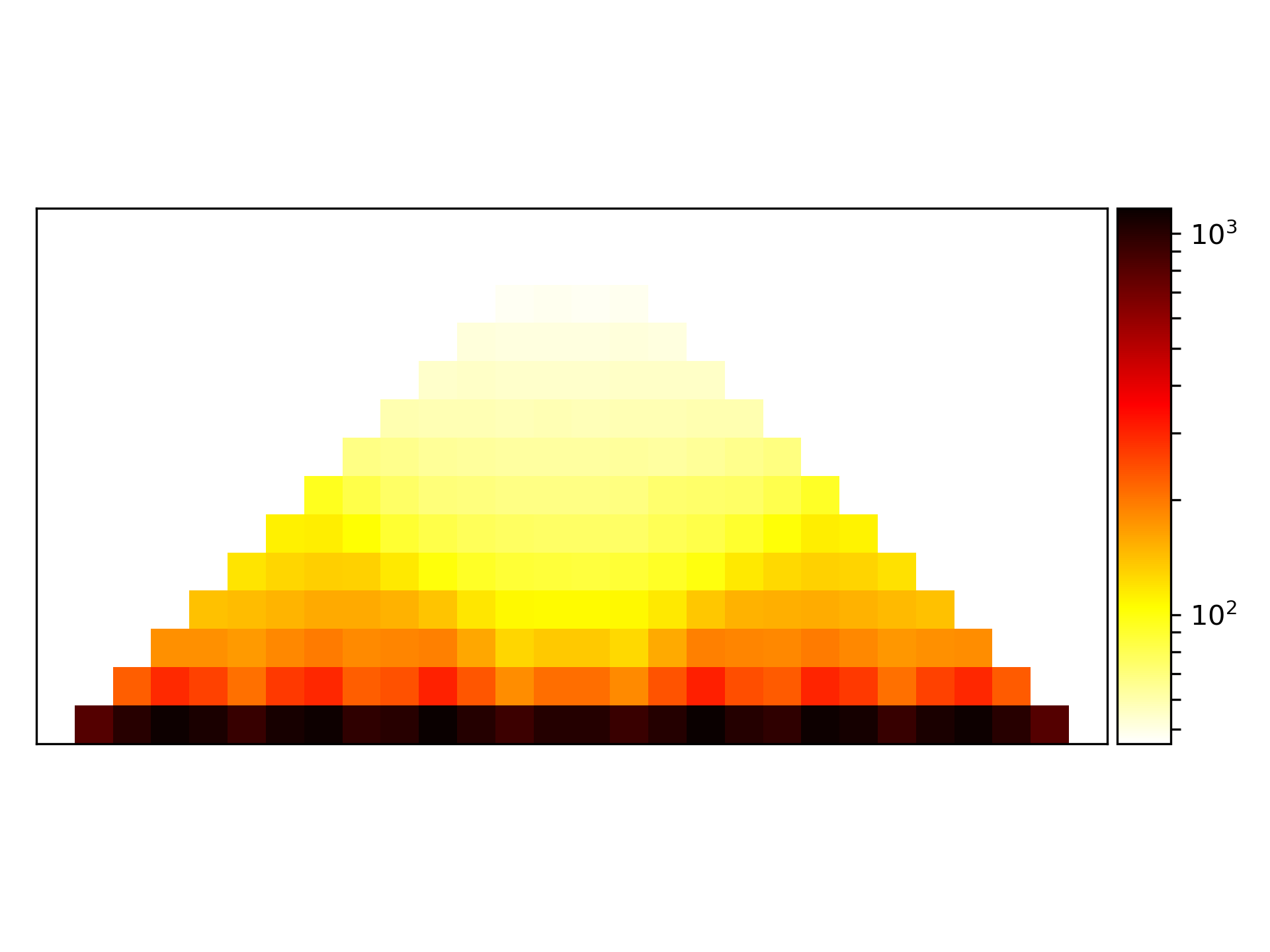
In a last step, the computed average region is plotted.
$ hicPlotAverageRegions -m primary_chr1.npz -o primary_chr1.png