Hi-C analysis of mouse ESCs using HiCExplorer¶
The following example shows how we can use HiCExplorer to analyze a published dataset. Here we are using a Hi-C dataset from Marks et. al. 2015, on mouse ESCs.
Protocol¶
The collection of the cells for Hi-C and the Hi-C sample preparation procedure was performed as previously described Lieberman-Aiden et al., with the slight modification that DpnII was used as restriction enzyme during initial digestion. Paired-end libraries were prepared according to Lieberman-Aiden et al. and sequenced on the NextSeq 500 platform using 2 × 75 bp sequencing.
Prepare for analysis¶
Download Raw fastq files¶
The fastq files can be downloaded from the EBI archive (or NCBI archive). We will store the files in the directory original_data.
mkdir original_data
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR195/007/SRR1956527/SRR1956527_1.fastq.gz -O original_data/SRR1956527_1.fastq.gz
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR195/007/SRR1956527/SRR1956527_2.fastq.gz -O original_data/SRR1956527_2.fastq.gz
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR195/008/SRR1956528/SRR1956528_1.fastq.gz -O original_data/SRR1956528_1.fastq.gz
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR195/008/SRR1956528/SRR1956528_2.fastq.gz -O original_data/SRR1956528_2.fastq.gz
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR195/009/SRR1956529/SRR1956529_1.fastq.gz -O original_data/SRR1956529_1.fastq.gz
wget ftp://ftp.sra.ebi.ac.uk/vol1/fastq/SRR195/009/SRR1956529/SRR1956529_2.fastq.gz -O original_data/SRR1956529_2.fastq.gz
Create an index¶
We start with creating an index for our alignment software for the GRCm38/mm10 genome. As a source we use the mm10 genome from UCSC
mkdir genome_mm10
wget http://hgdownload-test.cse.ucsc.edu/goldenPath/mm10/bigZips/chromFa.tar.gz -O genome_mm10/chromFa.tar.gz
tar -xvzf genome_mm10/chromFa.tar.gz
cat genome_mm10/*.fa > genome_mm10/mm10.fa
We have the mm10 genome stored in one fasta file and can build the index. We tried it successfully with hisat2, bowtie2 and bwa. Run the mapping with one of them and do not mix them!
hisat2¶
hisat2-build -p 8 genome_mm10/mm10.fa hisat2/mm10_index
You can find more information about hisat
bowtie2¶
bowtie2-build genome_mm10/mm10.fa bowtie2/mm10_index --threads 8
You can find more information about bowtie
bwa¶
bwa index -p bwa/mm10_index genome_mm10/mm10.fa
You can find more information about bwa
Mapping the RAW files¶
Mates have to be mapped individually to avoid mapper specific heuristics designed for standard paired-end libraries.
It is important to have in mind for the different mappers:
for either bowtie2 or hisat2 use the –reorder parameter which tells bowtie2 or hisat2 to output the sam files in the exact same order as in the .fastq files.
use local mapping, in contrast to end-to-end. A fraction of Hi-C reads are chimeric and will not map end-to-end thus, local mapping is important to increase the number of mapped reads.
Tune the aligner parameters to penalize deletions and insertions. This is important to avoid aligned reads with gaps if they happen to be chimeric.
hisat2¶
hisat2 -x hisat2/mm10_index --threads 8 -U ../original_data/SRR1956527_1.fastq.gz --reorder | samtools view -Shb - > SRR1956527_1.bam
hisat2 -x hisat2/mm10_index --threads 8 -U ../original_data/SRR1956527_2.fastq.gz --reorder | samtools view -Shb - > SRR1956527_2.bam
hisat2 -x hisat2/mm10_index --threads 8 -U ../original_data/SRR1956528_1.fastq.gz --reorder | samtools view -Shb - > SRR1956528_1.bam
hisat2 -x hisat2/mm10_index --threads 8 -U ../original_data/SRR1956528_2.fastq.gz --reorder | samtools view -Shb - > SRR1956528_2.bam
hisat2 -x hisat2/mm10_index --threads 8 -U ../original_data/SRR1956529_1.fastq.gz --reorder | samtools view -Shb - > SRR1956529_1.bam
hisat2 -x hisat2/mm10_index --threads 8 -U ../original_data/SRR1956529_2.fastq.gz --reorder | samtools view -Shb - > SRR1956529_2.bam
bowtie2¶
bowtie2 -x bowtie2/mm10_index --threads 8 -U ../original_data/SRR1956527_1.fastq.gz --reorder | samtools view -Shb - > SRR1956527_1.bam
bowtie2 -x bowtie2/mm10_index --threads 8 -U ../original_data/SRR1956527_2.fastq.gz --reorder | samtools view -Shb - > SRR1956527_2.bam
bowtie2 -x bowtie2/mm10_index --threads 8 -U ../original_data/SRR1956528_1.fastq.gz --reorder | samtools view -Shb - > SRR1956528_1.bam
bowtie2 -x bowtie2/mm10_index --threads 8 -U ../original_data/SRR1956528_2.fastq.gz --reorder | samtools view -Shb - > SRR1956528_2.bam
bowtie2 -x bowtie2/mm10_index --threads 8 -U ../original_data/SRR1956529_1.fastq.gz --reorder | samtools view -Shb - > SRR1956529_1.bam
bowtie2 -x bowtie2/mm10_index --threads 8 -U ../original_data/SRR1956529_2.fastq.gz --reorder | samtools view -Shb - > SRR1956529_2.bam
bwa mem -A 1 -B 4 -E 50 -L 0 -t 8 bwa/mm10_index original_data/SRR1956527_1.fastq.gz | samtools view -Shb - > SRR1956527_1.bam
bwa mem -A 1 -B 4 -E 50 -L 0 -t 8 bwa/mm10_index original_data/SRR1956527_2.fastq.gz | samtools view -Shb - > SRR1956527_2.bam
bwa mem -A 1 -B 4 -E 50 -L 0 -t 8 bwa/mm10_index original_data/SRR1956528_1.fastq.gz | samtools view -Shb - > SRR1956528_1.bam
bwa mem -A 1 -B 4 -E 50 -L 0 -t 8 bwa/mm10_index original_data/SRR1956528_2.fastq.gz | samtools view -Shb - > SRR1956528_2.bam
bwa mem -A 1 -B 4 -E 50 -L 0 -t 8 bwa/mm10_index original_data/SRR1956529_1.fastq.gz | samtools view -Shb - > SRR1956529_1.bam
bwa mem -A 1 -B 4 -E 50 -L 0 -t 8 bwa/mm10_index original_data/SRR1956529_2.fastq.gz | samtools view -Shb - > SRR1956529_2.bam
Build, visualize and correct Hi-C matrix¶
Create a Hi-C matrix using the aligned files¶
In the following we will create three Hi-C matrices and merge them to one.
Build Hi-C matrix¶
hicBuildMatrix builds the matrix of read counts over the bins in the genome, considering the sites around the given restriction site. We need to provide:
the input BAM/SAM files: –samFiles SRR1956527_1.sam SRR1956527_2.sam
binsize: –binSize 1000
restriction sequence: –restrictionSequence GATC
dangling end sequence: –danglingSequence GATC
the restriction sequence cut site file: create it with hicFindRestSite, the restriction sequence ‘GATC’ and the above used mouse genome fasta file
the name of output bam file which contains the accepted alignments: –outBam SRR1956527_ref.bam
name of output matrix file: –outFileName hicMatrix/SRR1956527_10kb.h5
the folder for the quality report: –QCfolder hicMatrix/SRR1956527_QC
the number of to be used threads. Minimum value is 3: –threads 8
the buffer size for each thread buffering inputBufferSize lines of each input BAM/SAM file: –inputBufferSize 400000
To build the Hi-C matrices:
mkdir hicMatrix
hicBuildMatrix --samFiles SRR1956527_1.bam SRR1956527_2.bam --binSize 10000 --restrictionSequence GATC --danglingSequence GATC --restrictionCutFile cut_sites.bed --outBam SRR1956527_ref.bam --outFileName hicMatrix/SRR1956527_10kb.h5 --QCfolder hicMatrix/SRR1956527_10kb_QC --threads 8 --inputBufferSize 400000
hicBuildMatrix --samFiles SRR1956528_1.bam SRR1956528_2.bam --binSize 10000 --restrictionSequence GATC --danglingSequence GATC --restrictionCutFile cut_sites.bed --outBam SRR1956528_ref.bam --outFileName hicMatrix/SRR1956528_10kb.h5 --QCfolder hicMatrix/SRR1956528_10kb_QC --threads 8 --inputBufferSize 400000
hicBuildMatrix --samFiles SRR1956529_1.bam SRR1956529_2.bam --binSize 10000 --restrictionSequence GATC --danglingSequence GATC --restrictionCutFile cut_sites.bed --outBam SRR1956529_ref.bam --outFileName hicMatrix/SRR1956529_10kb.h5 --QCfolder hicMatrix/SRR1956529_10kb_QC --threads 8 --inputBufferSize 400000
The output bam files show that we have around 34M, 54M and 58M selected reads for SRR1956527, SRR1956528 & SRR1956529, respectively. Normally 25% of the total reads are selected. The output matrices have counts for the genomic regions. The extension of output matrix files is .h5.
A quality report is created in e.g. hicMatrix/SRR1956527_10kb_QC, have a look at the report hicQC.html.
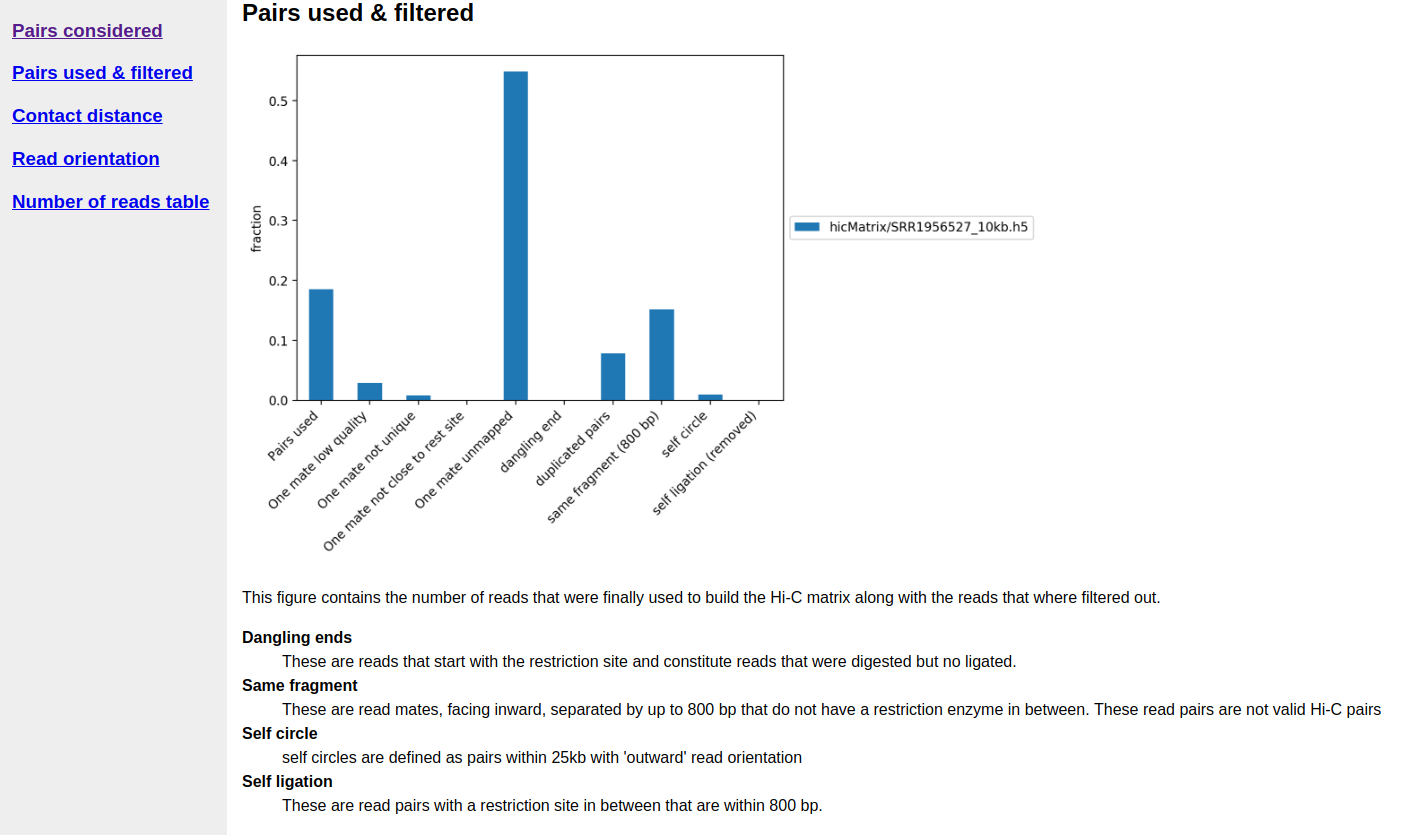
A segment of Hi-C quality report.¶
Merge (sum) matrices from replicates¶
To increase the depth of reads we merge the counts from these three replicates.
hicSumMatrices --matrices hicMatrix/SRR1956527_10kb.h5 hicMatrix/SRR1956528_10kb.h5 \
hicMatrix/SRR1956529_10kb.h5 --outFileName hicMatrix/replicateMerged_10kb.h5
Plot Hi-C matrix¶
A 10kb bin matrix is quite large to plot and is better to reduce the resolution (to know the size of a Hi-C matrix use the tool hicInfo), i.e. we usually run out of memory for a 1 kb or a 10 kb matrix and second, the time to plot is very long (minutes instead of seconds). For this we use the tool hicMergeMatrixBins.
Merge matrix bins for plotting¶
hicMergeMatrixBins merges the bins into larger bins of given number (specified by –numBins). We will merge 1000 bins in the original (uncorrected) matrix and then correct it. The new bin size is going to be 10.000 bp * 100 = 1.000.000 bp = 1 Mb
hicMergeMatrixBins \
--matrix hicMatrix/replicateMerged_10kb.h5 --numBins 100 \
--outFileName hicMatrix/replicateMerged.100bins.h5
Plot the corrected Hi-C matrix¶
hicPlotMatrix can plot the merged matrix. We use the following options:
the matrix to plot: –matrix hicMatrix/replicateMerged.100bins.h5
logarithmic values for plotting: –log1p
the resolution of the plot: –dpi 300
masked bins should not be plotted: –clearMaskedBins
the order of the chromosomes in the plot: –chromosomeOrder chr1 chr2 chr3 chr4 chr5 chr6 chr7 chr8 chr9 chr10 chr11 chr12 chr13 chr14 chr15 chr16 chr17 chr18 chr19 chrX chrY
the color map: –colorMap jet
the title of the plot: –title “Hi-C matrix for mESC”
the plot image itself: –outFileName plots/plot_1Mb_matrix.png
mkdir plots
hicPlotMatrix \
--matrix hicMatrix/replicateMerged.100bins.h5 \
--log1p \
--dpi 300 \
--clearMaskedBins \
--chromosomeOrder chr1 chr2 chr3 chr4 chr5 chr6 chr7 chr8 chr9 chr10 chr11 chr12 chr13 chr14 chr15 chr16 chr17 chr18 chr19 chrX chrY \
--colorMap jet \
--title "Hi-C matrix for mESC" \
--outFileName plots/plot_1Mb_matrix.png
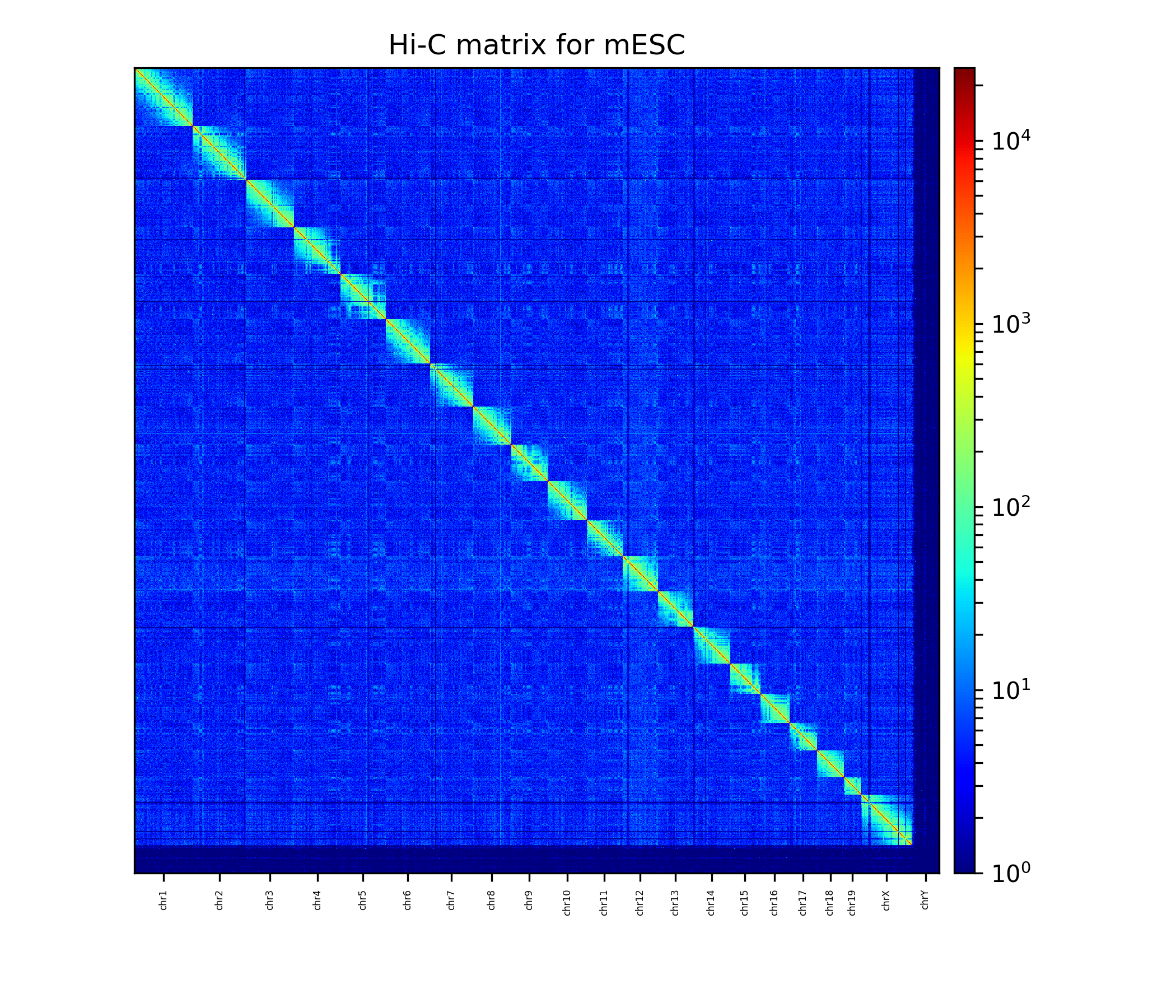
The Hi-C interaction matrix with a resolution of 1 MB.¶
Correct Hi-C Matrix¶
hicCorrectMatrix corrects the matrix counts in an iterative manner. For correcting the matrix, it’s important to remove the unassembled scaffolds (e.g. NT_) and keep only chromosomes, as scaffolds create problems with matrix correction. Therefore we use the chromosome names (1-19, X, Y) here. Important: Use ‘chr1 chr2 chr3 etc.’ if your genome index uses chromosome names with the ‘chr’ prefix.
Matrix correction works in two steps: first a histogram containing the sum of contact per bin (row sum) is produced. This plot needs to be inspected to decide the best threshold for removing bins with lower number of reads. The second steps removes the low scoring bins and does the correction.
In the following we will use a matrix with a bin size of 20 kb: 10kb * 2 = 20 kb
hicMergeMatrixBins \
--matrix hicMatrix/replicateMerged_10kb.h5 --numBins 2 \
--outFileName hicMatrix/replicateMerged.matrix_20kb.h5
(1-19, X, Y) variant:
hicCorrectMatrix diagnostic_plot \
--chromosomes 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 X Y \
--matrix hicMatrix/replicateMerged.matrix_20kb.h5 --plotName hicMatrix/diagnostic_plot.png
(chr1-ch19, chrX, chrY) variant:
hicCorrectMatrix diagnostic_plot \
--chromosomes chr1 chr2 chr3 chr4 chr5 chr6 chr7 chr8 chr9 chr10 chr11 chr12 chr13 chr14 chr15 chr16 chr17 chr18 chr19 chrX chrY \
--matrix hicMatrix/replicateMerged.matrix_20kb.h5 --plotName hicMatrix/diagnostic_plot.png
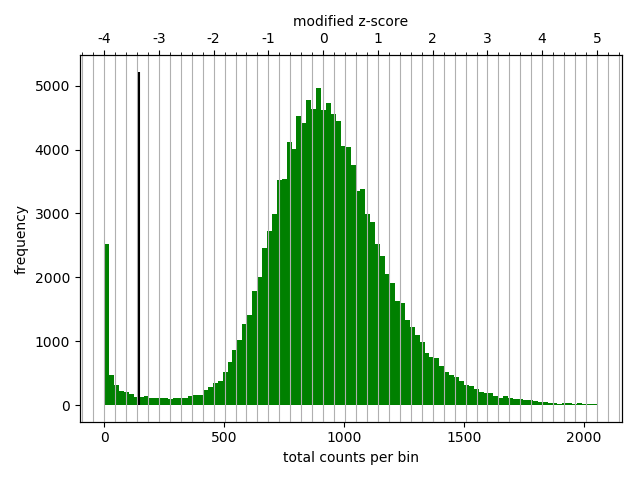
Diagnostic plot for the Hi-C matrix at a resolution of 20 kb¶
The output of the program prints a threshold suggestion that is usually accurate but is better to revise the histogram plot. The threshold is visualized in the plot as a black vertical line. See Example usage for an example and for more info.
- The threshold parameter needs two values:
low z-score
high z-score
“The absolute value of z represents the distance between the raw score and the population mean in units of the standard deviation. z is negative when the raw score is below the mean, positive when above.” (Source). For more information see wikipedia.

The z-score definition.¶
In our case the distribution describes the counts per bin of a genomic distance. To remove all bins with a z-score threshold less / more than X means to remove all bins which have less / more counts than X of mean of their specific distribution in units of the standard deviation.
Looking at the above distribution, we can select the value of -2 (lower end) and 3 (upper end) to remove. This is given by the –filterThreshold option in hicCorrectMatrix.
(1-19, X, Y) variant:
hicCorrectMatrix correct \
--chromosomes 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 X Y \
--matrix hicMatrix/replicateMerged.matrix_20kb.h5 \
--filterThreshold -2 3 --perchr --outFileName hicMatrix/replicateMerged.Corrected_20kb.h5
(chr1-ch19, chrX, chrY) variant:
hicCorrectMatrix correct \
--chromosomes chr1 chr2 chr3 chr4 chr5 chr6 chr7 chr8 chr9 chr10 chr11 chr12 chr13 chr14 chr15 chr16 chr17 chr18 chr19 chrX chrY \
--matrix hicMatrix/replicateMerged.matrix_20kb.h5 \
--filterThreshold -2 3 --perchr --outFileName hicMatrix/replicateMerged.Corrected_20kb.h5
It can happen that the correction stops with:
`ERROR:iterative correction:*Error* matrix correction produced extremely large values.
This is often caused by bins of low counts. Use a more stringent filtering of bins.`
This can be solved by a more stringent z-score values for the filter threshold or by a look at the plotted matrix. In our case we see that chromosome Y is having more or less 0 counts in its bins. This chromosome can be excluded from the correction by not defining it for the set of chromosomes that should be corrected, parameter –chromosomes.
In the case of multiple samples / replicates that need to be normalized to the same read coverage we recommend to compute first the normalization (with hicNormalize) and correct the data (with hicCorrectMatrix) in a second step.
Plot corrected matrix¶
We can now plot the one of the chromosomes (e.g. chromosome X) , with the corrected matrix.
- New parameter:
The region to plot: –region chrX:10000000-2000000 or –region chrX
(1-19, X, Y) variant:
hicPlotMatrix \
--log1p --dpi 300 \
-matrix hicMatrix/replicateMerged.Corrected_20kb.npz \
--region X --title "Corrected Hi-C matrix for mESC : chrX" \
--outFileName plots/replicateMerged_Corrected-20kb_plot-chrX.png
(chr1-ch19, chrX, chrY) variant:
hicPlotMatrix \
--log1p --dpi 300 \
--matrix hicMatrix/replicateMerged.Corrected_20kb.npz \
--region chrX --title "Corrected Hi-C matrix for mESC : chrX" \
--outFileName plots/replicateMerged_Corrected-20kb_plot-chrX.png
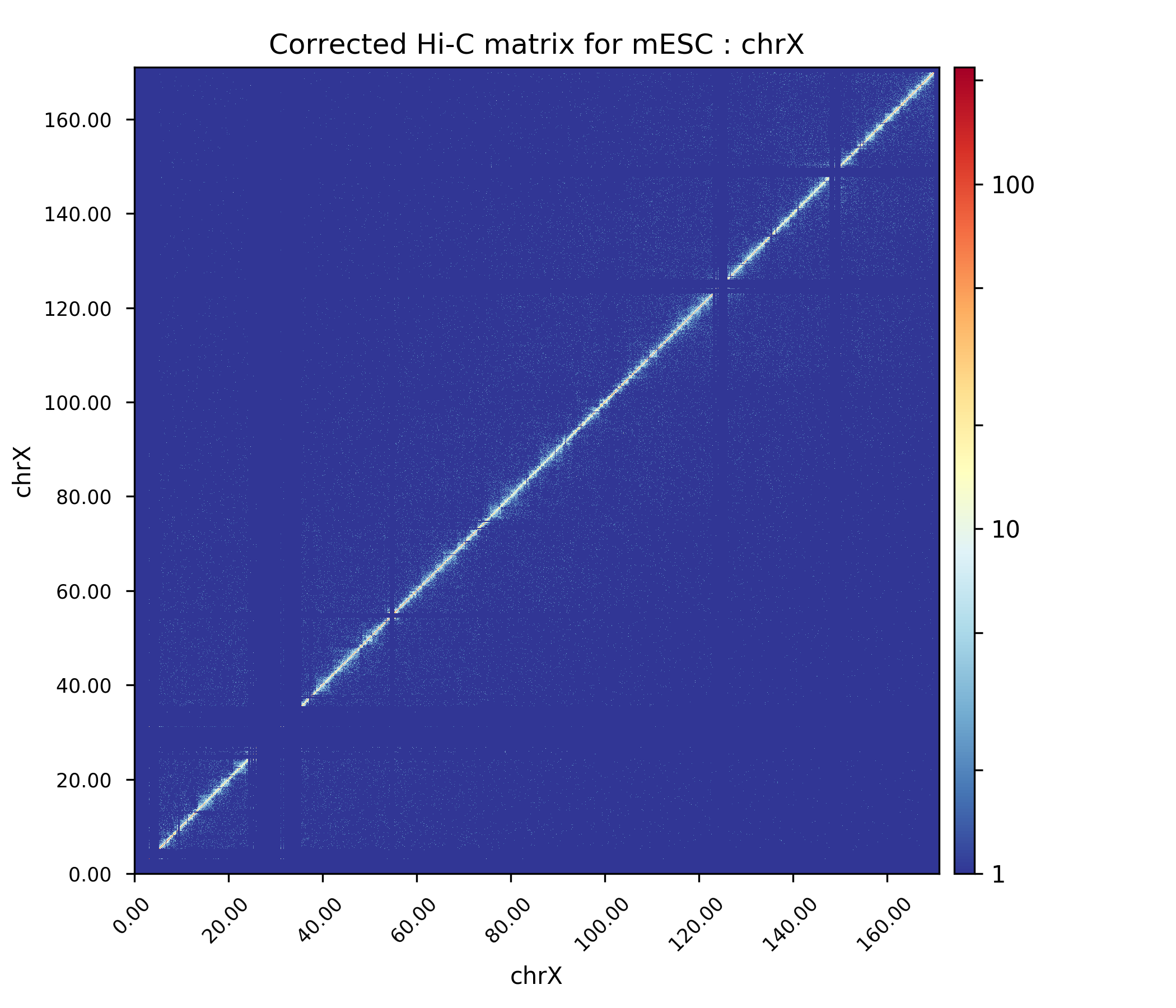
The Hi-C interaction matrix for chromosome X.¶
Plot TADs¶
“The partitioning of chromosomes into topologically associating domains (TADs) is an emerging concept that is reshaping our understanding of gene regulation in the context of physical organization of the genome” [Ramirez et al. 2017].
Find TADs¶
TAD calling works in two steps: First HiCExplorer computes a TAD-separation score based on a z-score matrix for all bins. Then those bins having a local minimum of the TAD-separation score are evaluated with respect to the surrounding bins to decide assign a p-value. Then a cutoff is applied to select the bins more likely to be TAD boundaries.
hicFindTADs tries to identify sensible parameters but those can be change to identify more stringent set of boundaries.
mkdir TADs
hicFindTADs --matrix hicMatrix/replicateMerged.Corrected_20kb.h5 \
--minDepth 60000 --maxDepth 120000 --numberOfProcessors 8 --step 20000 \
--outPrefix TADs/marks_et-al_TADs_20kb-Bins --minBoundaryDistance 80000 \
--correctForMultipleTesting fdr --threshold 0.05
As an output we get the boundaries, domains and scores separated files. We will use in the plot below only the TAD-score file.
Build Tracks File¶
We can plot the TADs for a given chromosomal region. For this we need to create a track file containing the instructions to build the plot. The hicPlotTADs documentation contains the instructions to build the track file.
In following plot we will use the listed track file. Please store it as track.ini.
[hic]
file = hicMatrix/replicateMerged.Corrected_20kb.h5
title = HiC mESC chrX:99974316-101359967
colormap = RdYlBu_r
depth = 2000000
height = 7
transform = log1p
file_type = hic_matrix
[tads]
file = TADs/marks_et-al_TADs_20kb-Bins_domains.bed
file_type = domains
border_color = black
color = none
line_width = 1.5
overlay_previous = share-y
show_data_range = no
[x-axis]
fontsize = 16
where = top
[tad score]
file = TADs/marks_et-al_TADs_20kb-Bins_score.bm
title = TAD separation score
height = 4
file_type = bedgraph_matrix
[spacer]
[gene track]
file = mm10_genes_sorted.bed
height = 10
title = mm10 genes
labels = false
We used as a gene track mm10 genes and sorted with sortBed from bedtools.
Plot¶
We plot the result with:
(1-19, X, Y) variant:
hicPlotTADs --tracks track.ini --region X:98000000-105000000 \
--dpi 300 --outFileName plots/marks_et-al_TADs.png \
--title "Marks et. al. TADs on X"
(chr1-ch19, chrX, chrY) variant:
hicPlotTADs --tracks track.ini --region chrX:98000000-105000000 \
--dpi 300 --outFileName plots/marks_et-al_TADs.png \
--title "Marks et. al. TADs on X"
The result is:
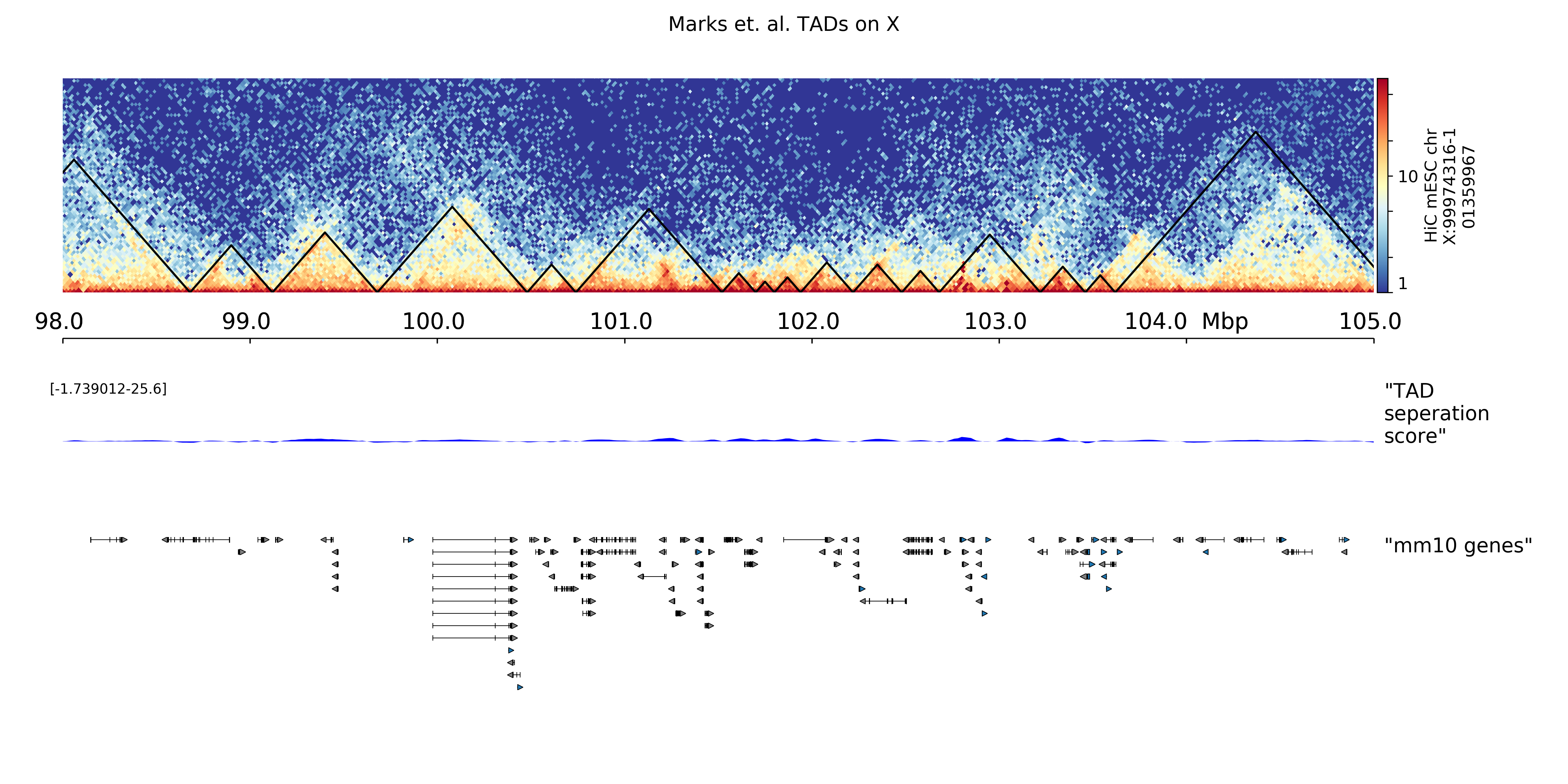
TADplot¶