hicValidateLocations¶
hicValidateLoops is a tool to compare the detect loops and TADs from hicDetectLoops / hicFindTADs (or from any other software as long as the data format is followed, see below) with known peak protein locations to validate if the computed loops / TADs have the expected anchor points. For example, loops in mammals are usually bound by CTCF or Cohesin, therefore it is important to know if the detect loops have protein peaks at their X and Y position.
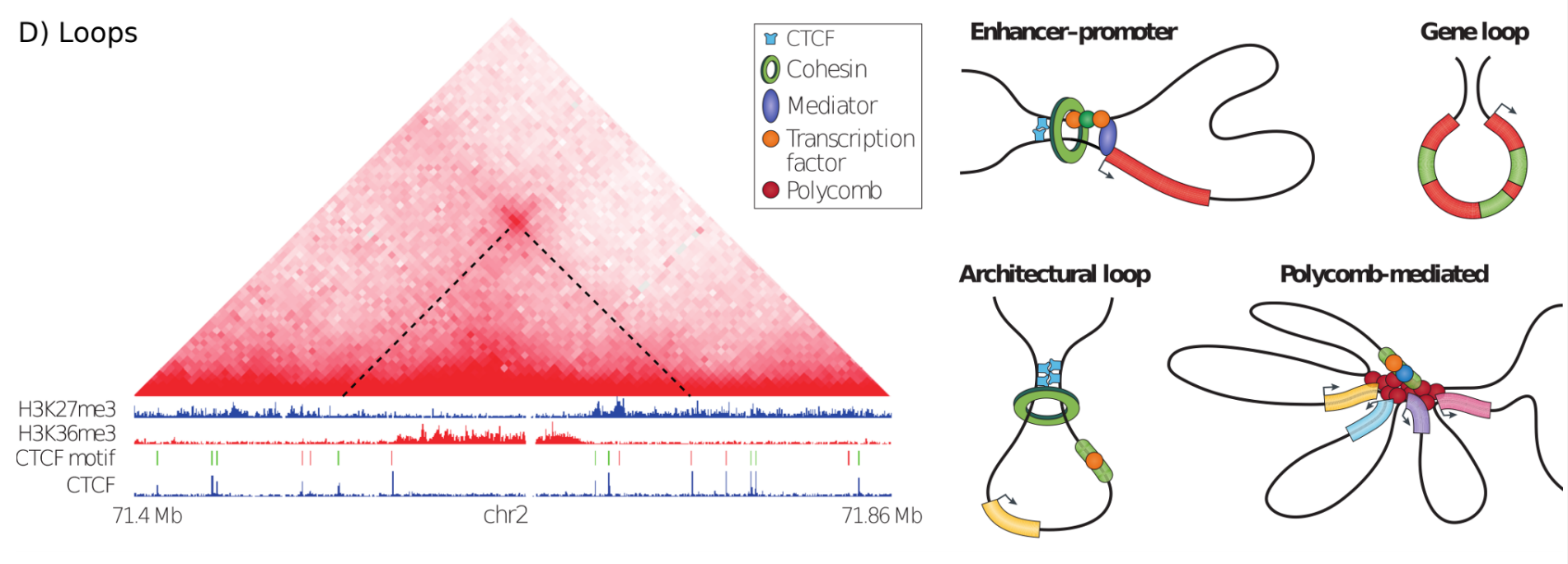
Loops in Hi-C, graphic from Bonev & Cavalli, Nature Reviews Genetics 2016¶
Data format¶
The data format of hicDetectLoops output is:
chr_x start_x end_x chr_y start_y end_y p-value
As --validationData the input of narrowPeak / broadPeak (both as bed) or a cool file is accepted. However, for the bed files, as long as the --validationData input file contains chromosome, start and end in the first three columns, it should work.
Concerning the TAD locations: Please use a file containing the boundary positions and not the domains!
This script overlaps the loop locations with protein locations to determine the accuracy of the loop detection. Loops need to have format as follows:
chr start end chr start end
The protein peaks need to be in narrowPeaks or broadPeak format.
A protein match is successfull if at the bin of the x and y location a protein peak is overlapped. A bin is assumed to have a protein if one or more protein peaks falling within the bin region. The value of the protein is not considered, only match or non-match.
usage: hicValidateLocations --data DATA --validationData VALIDATIONDATA
[--validationType {bed,cool}]
[--method {loops,tad}] --resolution RESOLUTION
[--outFileName OUTFILENAME]
[--chrPrefixLoops {None,add,remove}]
[--chrPrefixProtein {None,add,remove}] [--help]
[--version]
Required arguments¶
- --data, -d
The loop file from hicDetectLoops. To use files from other sources, please follow ‘chr start end chr start end’ format. For TAD data use the boundaries.bed file and not the domains file!
- --validationData, -vd
The data file to validate the given locations. Can be narrowPeak, broadPeak (both in bed), or cool
- --validationType, -vt
Possible choices: bed, cool
The type of the validation data. Can be bed, or cool format
Default: “bed”
- --method, -m
Possible choices: loops, tad
The method used (for the moment only loop is possible) (Default: “loops”).
Default: “loops”
- --resolution, -r
The used resolution of the Hi-C interaction matrix.
Optional arguments¶
- --outFileName, -o
The prefix name of the output files. Two file are written: output_matched_locations and output_statistics.First file contains all loop locations with protein location matches, second file contains statistics about this matching.
- --chrPrefixLoops, -cl
Possible choices: None, add, remove
Adding / removing / do nothing a ‘chr’-prefix to chromosome name of the loops.
- --chrPrefixProtein, -cp
Possible choices: None, add, remove
Adding / removing / do nothing a ‘chr’-prefix to chromosome name of the protein.
- --version
show program’s version number and exit