hicPlotDistVsCounts¶
Background¶
This tool allows a quick comparison between multiple Hi-C matrices of the Hi-C counts enrichment at different genomic ranges / distances up to whole chromosome. Biological replicates should display the exact same distribution while samples coming from different cell-lines, treated versus untreated samples or mutant versus wild-type samples should display a different distribution at long and/or close range.
The results of this tool usually reflect the proportion of long-range and short-range contacts calculated in each sample by hicQC. Local TAD or contact enrichments will not impact the results computed bu this tool, hicPCA is better suited for that purpose.
Description¶
This program creates distance vs. Hi-C counts plots. It can use several matrix files to compare them at once. If the –perchr option is given, each chromosome is plotted independently. When plotting multiple matrices, denser matrices are scaled down to match the sum of the smallest matrix.
usage: hicPlotDistVsCounts --matrices MATRICES [MATRICES ...] --plotFile file
name [--labels LABELS [LABELS ...]]
[--skipDiagonal] [--maxdepth INT bp] [--perchr]
[--chromosomeExclude CHROMOSOMEEXCLUDE [CHROMOSOMEEXCLUDE ...]]
[--outFileData OUTFILEDATA]
[--plotsize PLOTSIZE PLOTSIZE] [--help] [--version]
Required arguments¶
- --matrices, -m
Hi-C normalized (corrected) matrices. Each path should be separated by a space.
- --plotFile, -o
File name to save the file. The given file ending will be used to determine the image format. The available options are: .png, .emf, .eps, .pdf and .svg.
Optional arguments¶
- --labels
Label to assign to each matrix file. Each label should be separated by a space. Quote labels that contain spaces: E.g. –labels label1 “labels 2”. If no labels are given then the file name is used.
- --skipDiagonal, -s
If set, diagonal counts are not included.
Default: False
- --maxdepth
Maximum distance from diagonal to use. In other words, distances up to maxDeph are computed. Default is 3 million bp.
Default: 3000000
- --perchr
If given, computes and display distance versus Hi-C counts plots for each chromosome stored in the matrices passed to –matrices.
Default: False
- --chromosomeExclude
Exclude the given list of chromosomes. This is useful for example to exclude the Y chromosome. The names of the chromosomes should be separated by space.
- --outFileData
If given, the data underlying the plots is saved on this file.
- --plotsize
Width and height of the plot (in inches). Default is 6*number of cols, 4 * number of rows. The maximum number of rows is 4. Example: –plotsize 6 5
- --version
show program’s version number and exit
Usage example¶
hicPlotDistVsCounts should be used on corrected matrices with very large bins (e.g. at least 50kb bins), otherwise the curves will be spiky at longer ranges because of the sparness of the contacts, thus the likelyness of the samples will become hard to assess after a certain distance. hicPlotDistVsCounts is thus often ran after hicMergeMatrixBins and hicCorrectMatrix.
hicPlotDistVsCounts -m \
condition1_sample1_50_bins_merged.h5 \
condition1_sampel2_50_bins_merged.h5 \
condition2_sample1_50_bins_merged.h5 \
condition2_sample2_50_bins_merged.h5 \
-o counts_vs_dist_50_bins_merged.png \
--labels 'Cond 1 Sample 1' 'Cond 1 Sample 2' 'Cond 2 Sample 1' 'Cond 2 Sample 2' \
--maxdepth 20000000 \
--plotsize 5 4.2
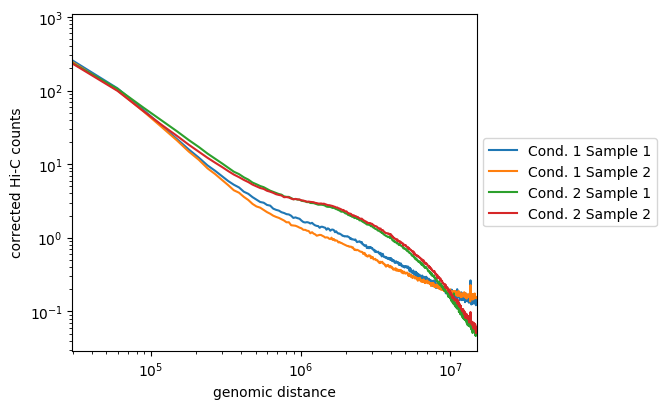
Here, we see that the samples from the first condition are not so well correlated, but they follow the same tendancies and are distinct from the two samples of the second condition. The later are well correlated and display enriched long-range contacts compared to the first condition samples.