hicCorrelate¶
Background¶
hicCorrelate is a dedicated Quality Control tool that allows the correlation of multiple Hi-C matrices at once with either a heatmap or scatterplots output.
Description¶
Computes pairwise correlations between Hi-C matrices data. The correlation is computed taking the values from each pair of matrices and discarding values that are zero in both matrices.Parameters that strongly affect correlations are bin size of the Hi-C matrices and the considered range. The smaller the bin size of the matrices, the finer differences you score. The –range parameter should be selected at a meaningful genomic scale according to, for example, the mean size of the TADs in the organism you work with.
usage: hicCorrelate --matrices MATRICES [MATRICES ...] [--zMin ZMIN]
[--zMax ZMAX] [--colorMap] [--plotFileFormat FILETYPE]
[--plotNumbers] [--method {pearson,spearman}] [--log1p]
[--labels sample1 sample2 [sample1 sample2 ...]]
[--range RANGE] --outFileNameHeatmap OUTFILENAMEHEATMAP
--outFileNameScatter OUTFILENAMESCATTER
[--chromosomes CHROMOSOMES [CHROMOSOMES ...]]
[--threads THREADS] [--help] [--version]
Required arguments¶
| –matrices, -m | Matrices to correlate (usually .h5 but other formats are allowed). hicCorrelate is better used on un-corrected matrices in order to exclude any changes introduced by the correction. |
Heatmap arguments¶
Options for generating the correlation heatmap
| –zMin, -min | Minimum value for the heatmap intensities. If not specified the value is set automatically. |
| –zMax, -max | Maximum value for the heatmap intensities.If not specified the value is set automatically. |
| –colorMap | Color map to use for the heatmap. Available values can be seen here: http://matplotlib.org/examples/color/colormaps_reference.html Default: “jet” |
| –plotFileFormat | |
Possible choices: png, pdf, svg, eps, emf Image format type. If given, this option overrides the image format based on the plotFile ending. The available options are: png, emf, eps, pdf and svg. | |
| –plotNumbers | If set, then the correlation number is plotted on top of the heatmap. Default: False |
Optional arguments¶
| –method | Possible choices: pearson, spearman Correlation method to use. Default: “pearson” |
| –log1p | If set, then the log1p of the matrix values is used. This parameter has no effect for Spearman correlations but changes the output of Pearson correlation and, for the scatter plot, if set, the visualization of the values is easier. Default: False |
| –labels, -l | User defined labels instead of default labels from file names. Multiple labels have to be separated by space, e.g. –labels sample1 sample2 sample3 |
| –range | In bp with the format low_range:high_range, for example 1000000:2000000. If –range is given only counts within this range are considered. The range should be adjusted to the size of interacting domains in the genome you are working with. |
| –outFileNameHeatmap, -oh | |
| File name to save the resulting heatmap plot. | |
| –outFileNameScatter, -os | |
| File name to save the resulting scatter plot. | |
| –chromosomes | List of chromosomes to be included in the correlation. |
| –threads | Number of threads. Using the python multiprocessing module. Is only used with ‘cool’ matrix format. One master process which is used to read the input file into the buffer and one process which is merging the output bam files of the processes into one output bam file. All other threads do the actual computation. Default: 4 |
| –version | show program’s version number and exit |
Usage example¶
Below, you can find a correlation example of uncorrected Hi-C matrices obtained from Drosophila melanogaster embryos, either wild-type or having one gene knocked-down by RNAi.
$ hicCorrelate -m Dmel_wt_1.h5 Dmel_wt_2.h5 Dmel_kd_1.h5 Dmel_kd_2.h5 \
--method=pearson --log1p \
--labels Dmel_wt_1 Dmel_wt_2 Dmel_kd_1 Dmel_kd_2 \
--range 5000:200000 \
--outFileNameHeatmap Dmel_heatmap --outFileNameScatter Dmel_scatterplot \
--plotFileFormat png
Heatmap¶
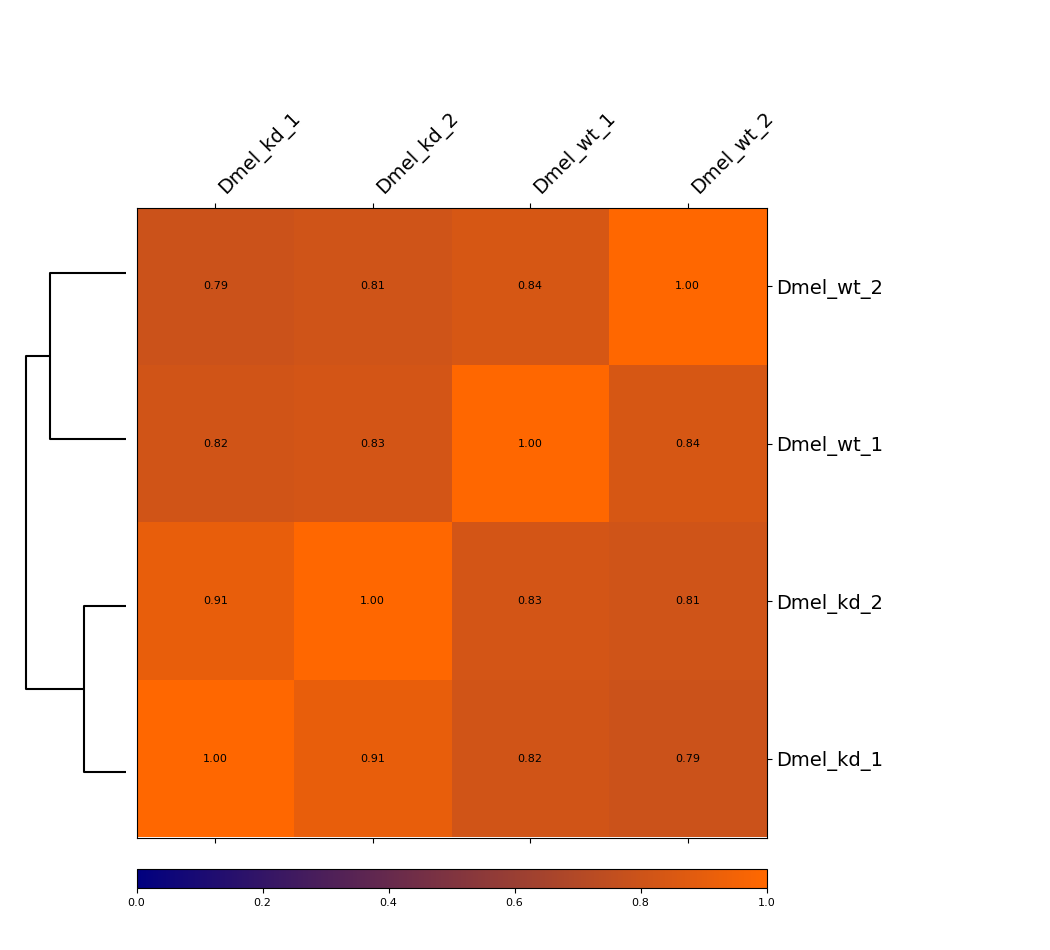
This example is showing a heatmap that was calculated using the Pearson correlation of un-corrected Hi-C matrices with a bin size of 6000 bp. The dendrogram indicates which samples are most similar to each other. You can see that the wild-type samples are seperated from the knock-down samples. The second option we offer is calculating the Spearman correlation.
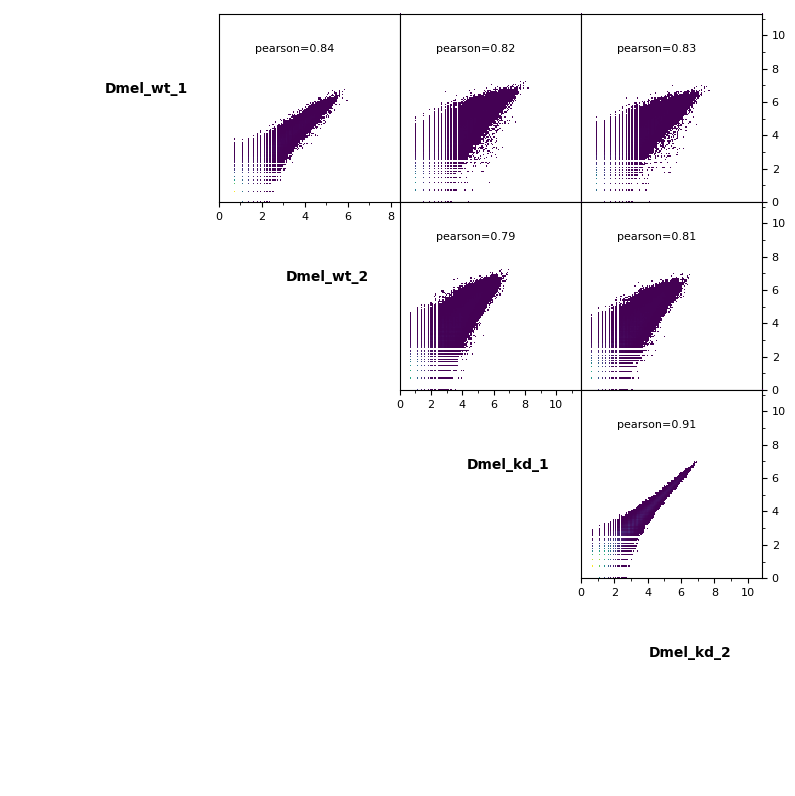
Scatterplot¶
Additionally, pairwise scatterplots comparing interactions between each sample can be plotted.