hicPlotTADs¶
Usage example¶
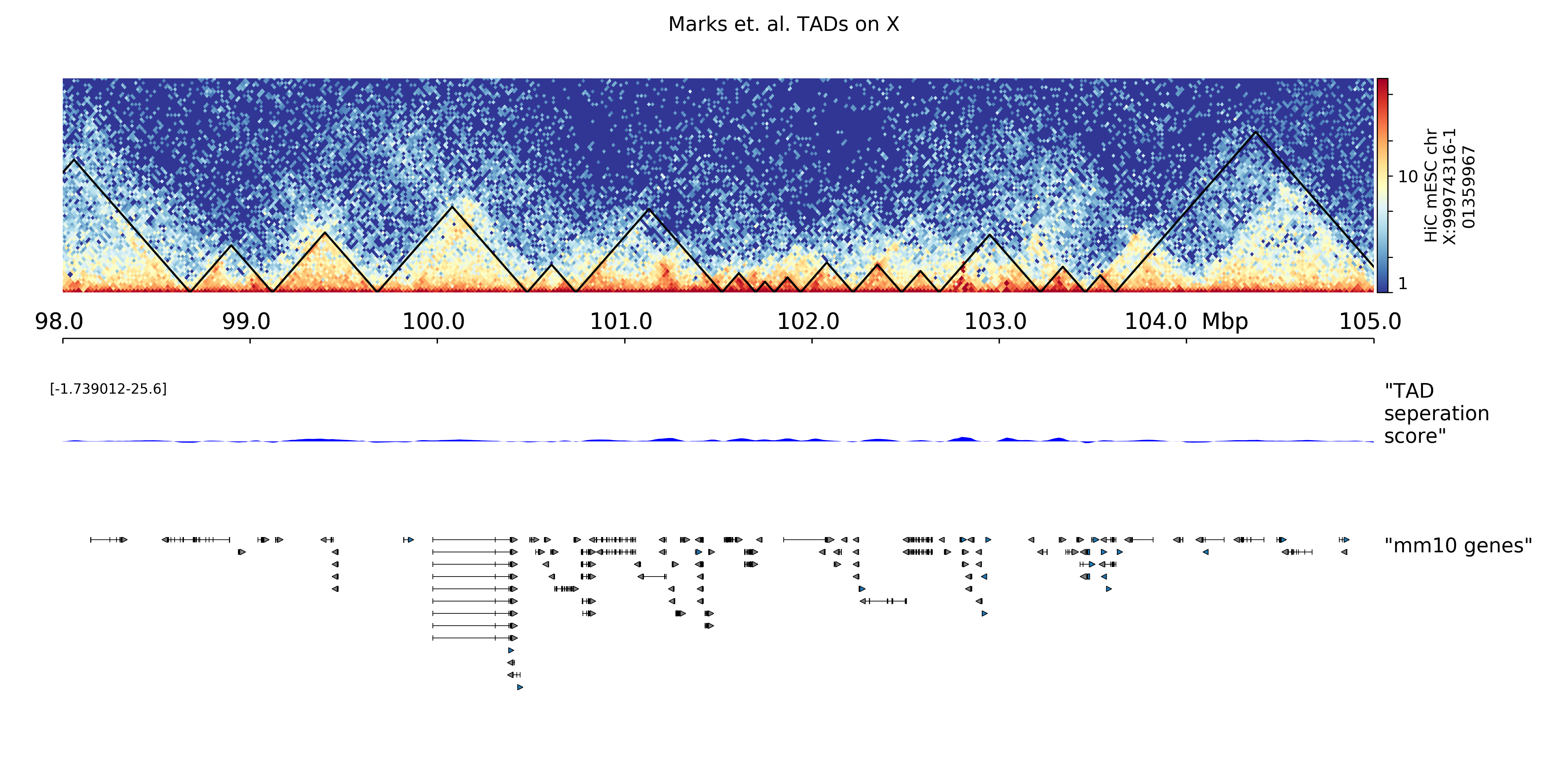
hicPlotTADs output is similar to a genome browser screen-shot that besides the usual genes, and score data (like bigwig or bedgraph files) also contains Hi-C data. The plot is composed of tracks that need to be specified in a configuration file. Once the tracks file is ready, hicPlotTADs can be used as follows:
$ hicPlotTADs --tracks tracks.ini --region chrX:99,974,316-101,359,967 \
-t 'Marks et. al. TADs on X' -o tads.pdf
Configuration file template¶
The following is a template for the configuration file which is based on .ini configuration files. Each track is defined by a section header (for example [hic track]), followed by parameters specific to the section as color, title, etc.
# lines that start with '#' are comment lines
# and are not interpreted by the program
# the different tracks are represented by sections in this file
# each section starts with a header of the form [hic]
# the content of the section label (in the previous example 'hic') is irrelevant for
# plotting and only used to tell the user when something goes wrong.
# There are two exceptions for this, the [x-axis] and the [spacer] sections
# that use the section label to determine the action.
[hic]
file = hic.h5
title = Hi-C
colormap = RdYlBu_r
depth = 100000
# optional arguments
min_value =2.8
max_value = 3.0
# transform options are log1p, log and -log
transform = log1p
boundaries_file = conductance_vs_hic/boundaries_all.bed
x labels = yes
type = interaction
# in case it can not be guessed by the file ending
file_type = hic_matrix
# show masked bins plots as white lines
# those bins that were not used during the correction
# the default is to extend neighboring bins to
# obtain an aesthetically pleasant output
show_masked_bins = yes
# optional, if the values in the matrix need to be scaled the
# following parameter can be used
scale factor = 1
[x-axis]
# optional
fontsize=20
# optional, options are top or bottom
where=top
# to insert a space simple add a
# section title [spacer]
[spacer]
#optional
width = 0.1
# You can also show the interactions as arcs between start and end bins.
# for this simply write the interactions in the Ginteraction format (HiCExport)
# and add the file here
[interactions]
file = Ginteractions.tsv
file_type = links
width = 10
color = black
title = HAS manual interactions
line width = 1
[bigwig]
file = file.bw
title = RNA-seq
color = black
width = 1.5
# optional values
min_value = 0
max_value = auto
# for each bin the average value is taken. The number of
# bins applies for the range being plotted. For example
# if 1Mb is plotted, then the average is computed for regions
# of 1000000/500 = 2000 bp
number of bins = 500
nans to zeros = True
# options are: line, points, fill. Default is fill
# to add the preferred line width or point size use:
# type = line:lw where lw (linewidth) is float
# similary points:ms sets the point size (markersize (ms) to the given float
type = line
# type = line:0.5
# type = points:0.5
# Default is yes, set to 'no' to turn off the visualization of
# text showing the data range (eg. 0 - 100) for the track
show data range = yes
# in case it can not be guessed by the file ending
# the file_type needs to be added
file_type = bigwig
[simple bed]
file = file.bed
title = peaks
color = read
# optional boder color. Set to none for no border color
border_color = black
width = 0.5
# optional. If not given is guessed from the file ending
file_type = bed
[genes]
# example of a genes track
# has the same options as a simple
# bed but if the type=genes is given
# the the file is interpreted as gene
# file. If the bed file contains the exon
# structure then this is plotted. Otherwise
# a region **with direction** is plotted.
file = genes.bed
title = genes
color = darkblue
width = 5
# optional
# to turn off/on printing of labels
labels = off
# options are 'genes' or 'domains'.
type = genes
# If not given is guessed from the file ending
file_type = bed
# optional: font size can be given if default are not good
fontsize = 10
[chrom states]
# this is a case of a bed file that is plotted 'collapsed'
# useful to plot chromatin states if the bed file contains
# the color to plot
file = chromatinStates.bed
title = chromatin states
# color is replaced by the color in the bed file
# in this case
color = black
# optional boder color. Set to none for no border color
border_color = black
# default behaviour when plotting intervals from a
# bed file is to 'expand' them such that they
# do not overlap. The display = collapsed
# directive overlaps the intervals.
display = collapsed
width = 0.3
[bedgraph]
file = file.bg
title = bedgraph track
color = green
width = 0.2
# optional. Default is yes, set to no to turn off the visualization of data range
show data range = yes
# optional, otherwise guessed from file ending
file_type = bedgraph
[bedgraph matrix]
# a bedgraph matrix file is like a bedgraph, except that per bin there
# are more than one value separated by tab: E.g.
# chrX 18279 40131 0.399113 0.364118 0.320857 0.274307
# chrX 40132 54262 0.479340 0.425471 0.366541 0.324736
# bedgraph matrices are produced by hicFindTADs
file = spectra_conductance.bm
title = conductance spectra
width = 1.5
orientation = inverted
min_value = 0.10
max_value = 0.70
# if type is set as lines, then the TAD score lines are drawn instead
# of the matrix
# set to lines if a heatmap representing the matrix
# is not wanted
type = lines
file_type = bedgraph_matrix
[vlines]
# vertical dotted lines from the top to the bottom of the figure
# can be drawn. For this a bed file is required
# but only the first two columns (chromosome name and start
# are used.
# vlines can also be given at the command line as a list
# of genomic positions. However, sometimes to give a file
# is more convenient in case many lines want to be plotted.
file = regions.bed
type = vlines